 >
> Perspective: Interaction Networks and AI for Structure-Based Drug Discovery

Many of our recent blog posts have dealt with physical modeling of small molecules and proteins. This is due to a recent flurry of groundbreaking research in equivariant neural networks that have improved structural modeling of chemicals. But today we step back to a broader-scope biochemistry problem. We are reviewing a paper from a few years ago, but one that I believe is worth returning to in the context of recent advances in structural biochemistry.
In 2018, Zitnix et. al. published “Modeling polypharmacy side effects with graph convolutional networks”. Drugs are often used in tandem in therapies for complex disease or for patients with co-existing conditions. These drug combinations, called “polypharmacy”, can be complementary or cause adverse side effects. But the span of all drug combinations is too large to test experimentally. Thus, the authors designed a neural network to predict polypharmacy side effects.
The Data
The training data is a relational graph, with nodes representing drugs or proteins and edges representing different types of relationships. Edges have different types, modeling protein-protein interactions, drug-protein interactions, and drug-drug side effects.
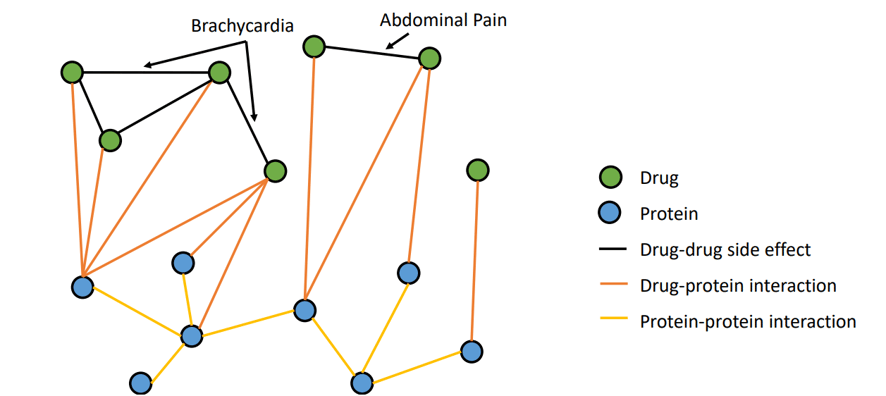
The Task
The model is trained with a cross entropy classification objective to predict whether different drug-drug edge types (representing side effects) are present. Because there are only positive examples of edges, the authors use negative sampling for negative examples. This involves random selection of other drug-drug edges which are assumed to be absent – a fair assumption due to the sparsity of the interaction network.
The Model
The authors use a graph convolutional network where node representations are updated based on their surrounding nodes. In this update, the surrounding nodes are transformed depending on their edge type, with a different learned projection matrix modeling each edge type. The node update is given in equation 1:
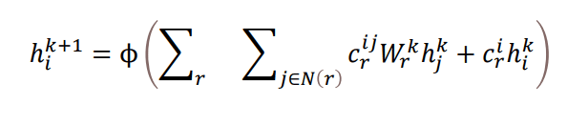
The result is updated representations for each node, which depend on interactions with nearby nodes in the graph.
Finally, the node representations are combined to predict edges. This is done via “Tensor factorization” – essentially, assuming that relationships can be modeled by a tensor which relates the two node embeddings. This is important, because it forces the prediction to depend on the combination of the two node embeddings and not on a single node.
The equations for drug-drug edge predictions, drug-protein edge predictions, and protein-protein edge predictions are given below, where is the predicted score.
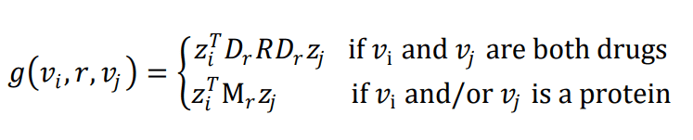
Takeaways
There are more takeaways from this paper than can be discussed here, but here are a few of mine:
- The use of tensor factorization to model multi-factor phenomena. If you want to model phenomena that depend on the interaction of two variables, tensor factorization is a good way to make sure that your model doesn’t ignore one variable.
- The consideration of broader biochemical effects in the drug discovery pipeline. This paper hints at a relatively unexplored side of AI for structure-based drug discovery – biochemical interaction networks. As structural modeling of small and large molecules improves, we can begin to fill in these sparse interaction networks. Experimental evidence will be replaced by relations discovered in-silico via structural drug-protein and protein-protein docking studies.
Using Structural Approaches to Construct Interaction Networks
The approach in this paper is creative but limited by the sparsity of the data and the lack of mechanistic understanding of disease. All the predicted edges are based on implicit understanding of drug-drug, drug-protein, and protein-protein interactions. Structural chemistry data gives mechanistic explanation of disease, and could offer generalization beyond existing drugs, since structure-based models explicitly capture the theorized physical interactions.
Structure-based interaction networks could lead to better structural understanding of disease symptoms, helping to identify new targets. They could shed light on mechanisms for drugs which are effective, but which have unknown mechanisms of action. They could be used to better predict ADMET properties, along with providing structural explanations.
Reducing the cost of drug discovery requires more than hit discovery and lead optimization, which have been the focus of AI for structure-based drug discovery. Using the recently developed structure-based tools to inform human-scale interaction networks is a difficult challenge but could unlock solutions to numerous other drug discovery problems. As with all AI driven research the proper handling of data is crucial, which is why we developed our life science data reservation platform to allow for the effective storing of data for use across multiple research teams and locations.


