 >
> How to identify active sites in enzymes with language models?

The European Lab for Learning and Intelligent Systems [ELLIS] recently held a workshop on “Machine Learning for Molecular Discovery” in conjunction with NeurIPS 2021. Highlights included papers leveraging physical symmetry to learn molecular force fields [1], information theoretic objectives to improve learning on molecular graphs [2], and more. All papers from the workshop can be found here.
In this blog post, we review “Identification of Enzymatic Active Sites with Unsupervised Language Modeling” by Kwate et. al. [3], a paper that achieves state-of-the-art unsupervised protein active site identification.
In drug discovery, labeling data is costly in terms of materials, researcher time, and potential for failure. For this reason, “unsupervised” methods which can leverage unlabeled data to extract information are especially useful.
Language modeling refers to machine learning on sequential data, using techniques originally developed for text, but which transfer well to protein sequences, SMILES molecular strings, etc. The authors used the popular “transformer” architecture for language modeling.
Transformers, in addition to achieving high accuracy on language modeling tasks, are structured in a way that allows for interpretability of what they learn. The fundamental building block of a transformer is an “attention mechanism”, in which the network assigns “attention values” to each part of the input and combines all inputs based on the attention value weights. By looking at the attention values, you can see which parts of the input were most important for classification. The authors used this attention value introspection technique to identify enzyme active sites.
Figure 1: Overview
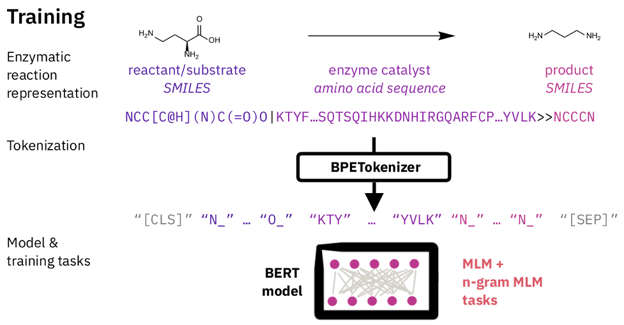
At training time, the authors used a masked language model [MLM] objective popularized by BERT [4]. They trained on 7 million reactions. The inputs were reaction reactants [SMILES, length r], enzyme catalyst [protein amino acid sequence, length m], and reaction products [SMILES, length p], concatenated together. They then masked [or blanked] tokens in the input, and asked the network to predict the identity of the missing token. The idea is that in the process of learning this unsupervised task, the network will use information from the protein active site to predict the missing ligand tokens. We can then examine the attention values of the network to see which parts of the protein helped to predict the ligand tokens, and assign those to be the active site.
The active site extraction procedure is as follows:
- From the trained language model, they extract the [r + m + p] x [r + m + p] attention matrix, A.
- From A, they extract the parts that allow cross-attention between the reactant and the enzyme [two r x m sub-matrices] and add them together to get an [r x m] matrix P.
- Each atom in the reactants votes for the k top amino acid matches by taking the k maximum values in its row of the matrix P.
- Each voted-for amino acid is predicted to be part of the active site.
The authors’ technique, which they called “RXNAAMapper”, achieved an “overlap score” 7 absolute percentage points higher than the existing Pfam method, while achieving a false positive rate 12 absolute percentage points lower – a significant improvement.
Figure 2: Left, Pfam predictions. Right, RKNAAMapper.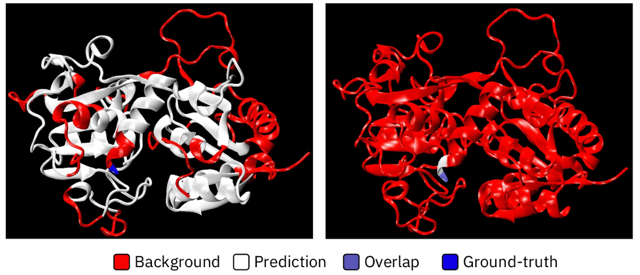
What are some takeaways from this paper? The authors used cheaper, more abundant, lower-resolution reaction data combined with unsupervised learning to infer structural information that would otherwise require expensive, high-resolution crystallography experiments. The inventors of AlphaFold 2 [5] used a similar technique to leverage 300 million+ proteins without experimental structures to help with the prediction of 3D protein structures.
This can be applied to other settings – maybe you have low-resolution mass-spectrometry data for a soup of molecules and want to infer an interaction network. Can you conceive an unsupervised training scheme that would allow you to determine which molecules interact?
Sources:
- Learning Small Molecule Energies and Interatomic Forces with an Equivariant Transformer on the ANI-1x Dataset; Bryce Hedelius, Fabian Bernd Fuchs, Dennis Della Corte
- 3D Infomax improves GNNs for Molecular Property Prediction; Hannes Stärk, Dominique Beaini, Gabriele Corso, Prudencio Tossou, Christian Dallago, Stephan Günnemann, Pietro Lio
- Identification of Enzymatic Active Sites with Unsupervised Language Modeling; Loïc Dassi Kwate, Matteo Manica, Daniel Probst, Philippe Schwaller, Yves Gaetan Nana Teukam, Teodoro Laino
- BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding; Jacob Devlin, Ming-Wei Chang, Kenton Lee, Kristina Toutanova
- Highly accurate protein structure prediction with AlphaFold; John Jumper et. al.


