 >
> The Protein Folding Problem – is it Solved?
In CASP14, DeepMind presented the results of AlphaFold, a deep neural network designed for protein structure prediction. During the experiment, AlphaFold predicted structures with an average deviation of ~1 Å from the C-alpha atoms of experimentally solved structures. Now, ~1 Å is often also used as the resolution to denote high accuracy experimental structures (they come in a range of 1 Å – 20 Å, depending on protein and experiment). However, root mean square deviation (RMSD) of 1 Å as measured during CASP is not the same as 1 Å experimental resolution click here for details. Because these values are so similar, many declared the protein folding problem to be solved.
However, in a recent letter to Science, Peter B. Moore et. al. argued that the well-publicized success of AlphaFold is not all it seems. In part 1 of this 2-part blog, we address two of the four key points of their argument, resolution accuracy and dynamic structure variation. Part 2 of this blog will address validation and if protein folding is “solved”.
Resolution Accuracy
“AlphaFold achieves a mean C-alpha root mean square deviation root mean square deviation (RMSD) accuracy of ∼1 Å for the Critical Assessment of Structure Prediction 14 CASP14 dataset… At present, for the best cases, the C-alpha coordinate RMSD accuracy of AlphaFold-predicted structures roughly corresponds to the accuracy expected for structures determined at resolutions no better than ∼4 Å.”
AlphaFold measures its success by C-alpha RMSD, reporting an accuracy of ∼1 Å. However, this should not be compared directly to the resolution reported by experimental methods like x-ray crystallography and cryo-EM. In experimental methods, a 1 Å resolution applies to all atoms, not just C-alpha atoms. Most protein interactions that are relevant to human biochemistry and drug design are based on interactions with protein side chains, requiring high accuracy for side chains, not just C-alpha coordinates. You can see an example of this in figure 1, which compares a 1 Å structure of lysosyme PDB 5HMV with a 4 Å structure of lysosyme PDB 1BHZ. The 4 Å structure has a reasonably accurate backbone, but the side chains are low inaccuracy. High accuracy for the full, atomistic structure is necessary for understanding protein-protein and protein-drug interactions.
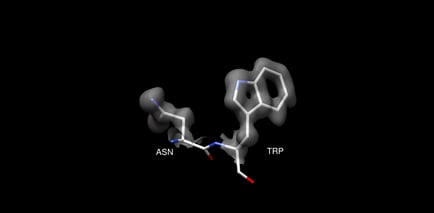
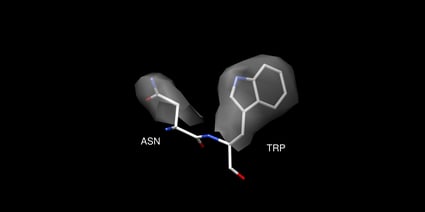
Top: Backbone of 5HMV blue and 1BHZ tan. Bottom: Backbone and side chains of 5HMV blue and 1BHZ tan. The low-resolution structure matches the correct backbone but fails to accurately predict side chains see misaligned side chains at the top of the aligned structures.
Dynamic Structure Variation
“A further complication for structure prediction is the dynamic structural variation in a given sequence. Allosteric states, which can differ dramatically, may be in an intrinsic equilibrium or depend on a binding partner…”
AlphaFold is trained on static protein structures in the Protein Data Bank [2]. These static structures do not account for the inherent dynamic motion of the protein, nor do they account for the interactions with binding partners, which can be small molecules, proteins, or other ligands.
You can see an example of this in figure 2, an example from the paper, “Protein Conformational Changes Are Detected and Resolved Site Specifically by Second-Harmonic Generation. These are two structures of the same protein, but the protein adopts a significant conformational change upon binding maltose.
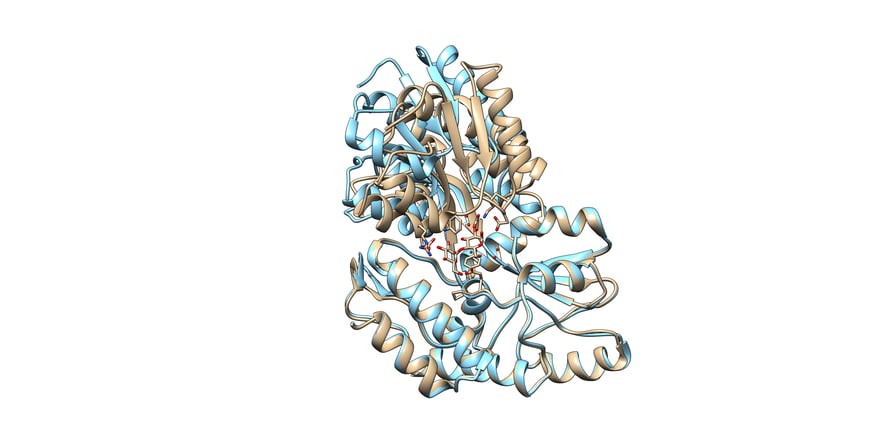
Two aligned structures of MBP, maltose-binding protein, with maltose bound tan, PDB 1ANF and unbound blue, PDB 1JW4.
In a recent letter to Science, Peter B. Moore et. al. argued that the well-publicized success of AlphaFold is not all it seems. In Part 1 of this 2-part blog, we addressed resolution accuracy and dynamic variation points of their argument. In Part 2, we address the final two points, validation and if protein folding is “solved”.
Validation
“Recent advances should be taken as a call for further development. Moreover, lessons should be learned from history. In 1990, Alwyn Jones and Carl-Ivar Brändén published a commentary on errors in x-ray crystal structures that stimulated the development of cross-validation and validation tools for structural biology and that ultimately made the databases of experimental structures much more reliable.”
As with other tools, AlphaFold is designed to perform well on the benchmarks on which it is evaluated, and is subject to bias based on its training data. We should seek out the failure modes of AlphaFold and develop new metrics as starting points for improvement. Jones and Brändén identified 7 possible sources for errors in crystal structures. In the ensuing two decades, a slough of crystal structure validation tools became available, including checks for bond lengths and angles, checks for the planarity of peptide bonds, and more – see A New Generation of Crystallographic Validation Tools for the Protein Data Bank. Now, each structure published on the PDB comes with validation scores indicating the quality of the structure. Similar approaches should be used to improve validation of AlphaFold-predicted structures.
“Solving” Protein Folding
“Finally, it is necessary to reflect on what the word “solved” might mean in the context of the protein-folding problem… we feel that solving the protein-folding problem means making accurate predictions of structures from amino acid sequences starting from first principles based on the underlying physics and chemistry.”
AlphaFold is trained on a dataset that is a subset of all protein structures that exist. In the authors’ opinion, “solving” the protein folding problem means discovering the underlying principles that govern protein folding, and making accurate predictions based on those principles. First-principles approaches are believed to generalize better than parameterized models trained on data.
These are valid criticisms and offer opportunity for improvement. In particular, the argument for first-principles models is compelling – though I would argue that much of the success of AlphaFold is in fact due to incorporating first-principles. Unlike previous protein folding neural networks, AlphaFold used recurrent, equivariant operations on atomic coordinates, simulating evolution of the atomic system forward in time. Other works, like the recent “EquiDock”, build other known physics principles such as non-intersection of molecules into their models. As researchers incorporate more physics principles into their neural models, we can expect those models to achieve higher accuracy with fewer data. With so much data available to scientists and researchers from different fields and locations it’s increasingly important that this data be securely preserved and accessible making a LIMS consolidation solution an essential part to modern researchers.


