 >
> SELFIES and the Future of Molecular String Representations

Neural sequence models have recently produced astonishing results in domains ranging from natural language to proteins and biochemistry. Current sequence models trained on text can explain jokes, answer trivia, and even write code. AlphaFold is a sequence model trained to predict protein structure with near-experimental accuracy.
In the chemistry domain, sequence models have also been used for learning problems on small, drug-like molecules. However, the most common syntax for representing small molecules as sequences runs into syntactic errors, limiting the usefulness of neural sequence models for generating new small molecules. In response, Krenn et. al. developed a new syntax for representing small molecules called SELFIES, for which all strings represent valid molecules. This makes it possible to leverage the power of neural sequence models to generate new molecules, with promising implications for drug discovery.
In this post, we review not the original SELFIES paper, but a review paper from 31 researchers across 29 institutions, advocating the use of SELFIES and outlining opportunities and open problems related to this research direction.
The authors present 16 different research directions, but we’ll focus on those most relevant to drug discovery.
Benchmarks for Generative Molecular Design
One research direction the authors propose is to create new benchmarks for generative molecular design. Benchmarks for generative molecular design currently involve distribution-matching or goal-oriented molecular design. However, current methods achieve perfect scores on these benchmarks, presenting the need for more difficult benchmarks to evaluate current models. These benchmarks might involve evaluation of ADMET properties, synthesizability, and protein binding affinity.
Smoothness of Generated Molecules with Respect to SELFIES-Based Representations
Representation learning is the task of learning vector representations of data to reduce noise and make downstream learning problems easier. With good representations, you can interpolate between the vector representations of two molecules, and the molecules corresponding to the interpolated vectors should transition smoothly from one molecule into the other. Previous approaches to representation learning of molecules have trained VAEs on SMILES molecular strings, but since not all representation vectors corresponded to valid SMILES strings, it was difficult to measure the smoothness of the transitions between molecules.
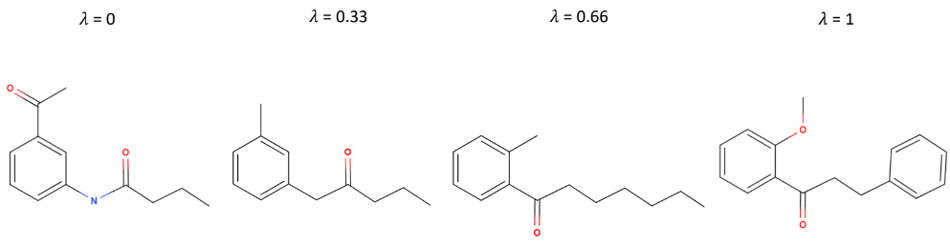
If a is the vector representing the first molecule, b is the vector representing the last, and the vector representing each intermediate molecule is a + (1-λ)b , SELFIES strings that are smooth with respect to the latent space might correspond to something like the molecules above, where structures smoothly transition into each other.
Smoothness of Molecular Properties with Respect to SELFIES-Based Representations
In representation learning, the space of all representations is called the “latent space”. Similarly to the previous point, it would be helpful to know if the latent space is organized such that there is a smooth relationship between latent representation and molecular properties.
Why does smoothness matter? If molecules and molecular properties are smooth with respect to representations, we can apply gradient-based techniques to find a vector in the latent space that maximizes certain desirable molecular properties, and then translate that vector into a molecule with those properties.
Learning What the Machine has Learned in the Latent Space
It is common to see visualizations of a neatly organized latent space, though the actual smoothness could be quite different. Understanding the proximity of different molecules in the latent space is an important step in answering questions about smoothness. It could also provide insight about the structural motifs that lead to certain drug-like properties, aiding in new hypotheses about drug-like molecules.
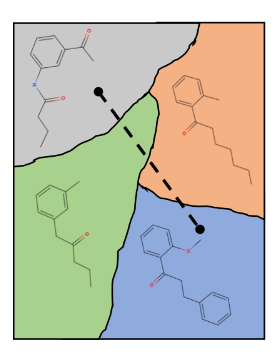
An example visualization of a neatly organized 2-dimensional latent space, with smooth transitions between molecules.
Applications of SELFIES
These are all ideas that are focused on research that builds on SELFIES, not necessarily including applications of SELFIES. However, SELFIES provide opportunities for improved models for many applications.
Chemical reaction modeling could be aided by the use of SELFIES. In reaction prediction, a product is predicted from the reactants, agents, and conditions. In retrosynthesis, reactants are predicted from the product. In reaction property prediction, the entire reaction is given and yield, energy profile, or other properties are predicted.
SELFIES also lend themselves to generative modeling of drugs. Given a protein active site, you could train a model to generate potential binders in SELFIES format.
Robust molecular string representations could help in all these tasks. They eliminate the problem of hack-ish proposal-rejection methods for generating molecular strings. If you are using a neural sequence model for small molecules, you should consider using the SELFIES representation. Additionally you should consider using a solution like ZONTAL’s life science data preservation platform to securely store your models data.


