 >
> MILCDock – Machine Learning Consensus Docking

Molecular docking tools are commonly used in drug discovery to computationally identify new molecules through virtual screening. However, these tools often suffer from inaccurate scoring functions that can vary in performance across different proteins. To address this issue, researchers at Brigham Young University have developed MILCDock, a machine learning consensus docking tool that uses predictions from five traditional molecular docking tools to improve the accuracy of binding affinity predictions.
In MILCDock, features are extracted from the output of the five traditional docking tools and provided as input to a neural network. The network then predicts the probability that a given ligand will bind to a protein based on these features. The model architecture consists of an ensemble of feed-forward neural networks, which outputs the probability of binding.
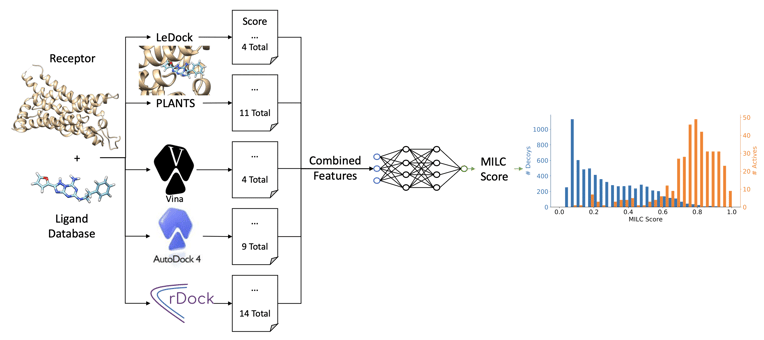
Figure 1: The ML model architecture used in MILCDock. The model takes features extracted from traditional docking tools as input and outputs a probability that a ligand will bind to a protein.
MILCDock was trained and tested on data from both the DUD-E and LIT-PCBA docking datasets. The DUD-E and LIT-PCBA datasets are commonly used for evaluating molecular docking methods. The DUD-E dataset consists of 102 protein targets associated with libraries of small molecules labeled as actives or decoys against their target. The LIT-PCBA dataset consists of 15 protein targets and a set of ~200,000 in vitro-tested compounds labeled as active or inactive.
An important difference between the two datasets is the type of compounds they contain. The DUD-E dataset contains active and decoy compounds, whereas the negatively labeled compounds in the LIT-PCBA dataset are experimentally verified as inactive. Decoy compounds are chemically similar to active compounds but are not experimentally verified.
The results showed improved performance over traditional molecular docking tools and other consensus docking methods on the DUD-E dataset. The LIT-PCBA targets proved to be difficult for all methods tested.
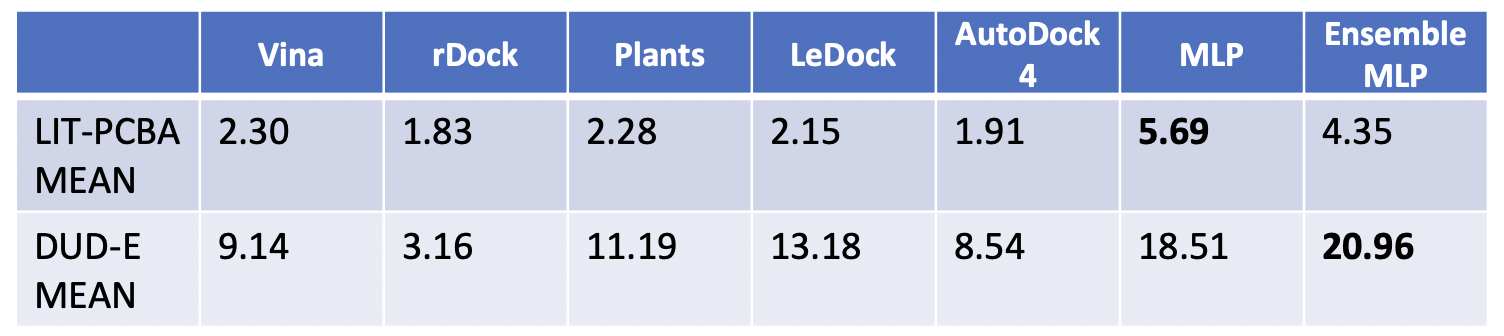
Figure 2: Comparison of MILCDock to traditional docking methods on the DUD-E and LIT-PCBA datasets. MILCDock shows improved performance over traditional methods on the DUD-E dataset, but performance is more mixed on the LIT-PCBA dataset.
The researchers also found that DUD-E data, although biased, can be effective in training machine learning tools if care is taken to avoid the biases during training. This is an important consideration when using machine learning for virtual screening, as biases in the training data can lead to inaccurate predictions and hinder the success of drug discovery efforts.
Overall, MILCDock represents a significant advance in the use of machine learning for consensus docking, improving the accuracy and robustness of binding affinity predictions and enabling more efficient and effective virtual screening in drug discovery.


