 >
> Deep Learning for Designing Protein Scaffolds
In a recent preprint from the Baker Lab, Jue Wang et. al. outlined a framework for protein design that uses protein structure prediction neural networks. This framework defines several types of protein design problems and demonstrates multiple effective approaches to solve these problems. Here we condense this detailed study into its main points.
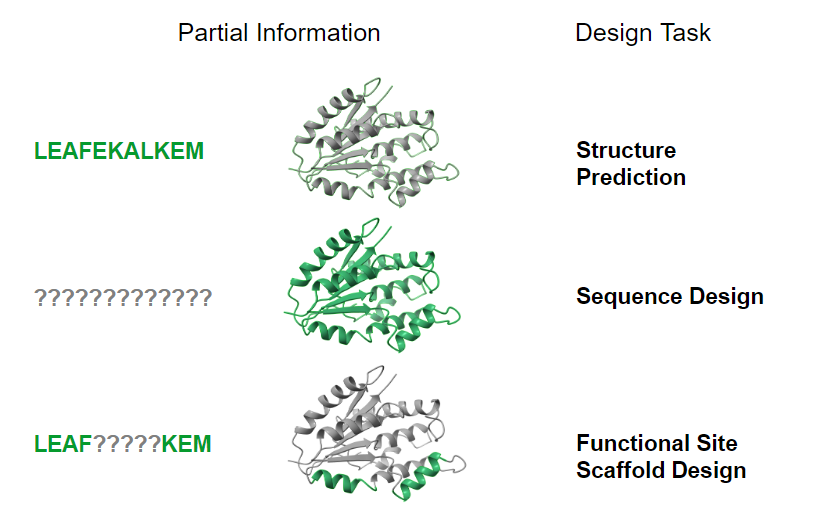
Protein Design Tasks
The authors identify several possible protein design tasks with different information provided and missing. In this paper, they focus on the fourth task, functional site scaffold design. In this problem, a “motif”, or functional site, is provided, with known residue identities and coordinates. The task is to predict a scaffold – a protein backbone that is stable and which contains the motif.
Two Complimentary Methods
The authors use two main approaches to design functional sites: “constrained hallucination” and “information recovery” aka, “inpainting”.
Constrained hallucination consists of taking an already-trained protein structure prediction network like trRosetta, RosettaFold, or AlphaFold, and optimizing over the input sequence to minimize a loss. The loss consists of a “motif loss”, which incentivizes certain residues to match a desired target structure, and a “hallucination loss” which incentivizes the protein to reach a well-folded state. The constrained hallucination method works well for proteins with very little information provided i.e. only the functional site but is slow, due to an optimization procedure that must be performed for each protein that is designed.
On the other hand, the information recovery method is faster, requiring only a forward pass through the structure prediction network, and works well for proteins where very little information is missing, e.g. the scaffold is provided but the functional site is missing. The information recovery method works by taking as input a masked sequence, and predicting the residue identities and 3D coordinates of the masked part of the sequence. This feature is built in to the RosettaFold architecture, and the authors trained a version of that architecture to excel at this inpainting task.
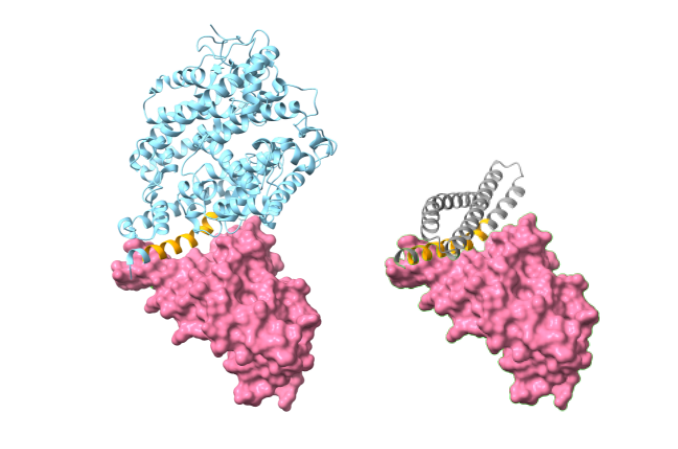
Three Applications
The functional site scaffold design problem is best suited to the first approach, so the authors applied the constrained hallucination approach to three different functional site design problems. In the first case, they designed a “receptor trap”, a protein that blocks the binding site of a viral protein. In the second experiment, they designed a scaffold around a metal binding site. In the third, they designed a protein-protein interface as a scaffold around a known binding region.
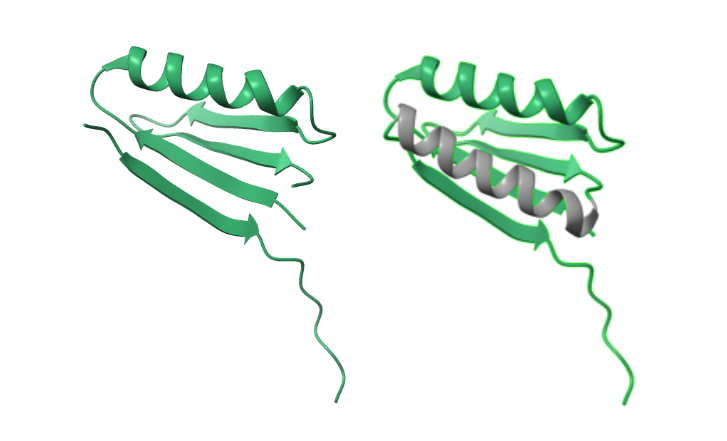
Caption: Three applications tested by the authors. In yellow is the original protein. In orange is the target motif. In purple is the predicted motif. In gray is the predicted scaffold. Top Left: Receptor trap design. Top Right: Metal binding site design. Bottom: Protein interface design.
Inpainting
The authors also experimented with the inpainting model, and found that it was able to recover masked sequences. They were able to generate more varied “inpainted” proteins by providing less information about surrounding residues.
AlphaFold Validation
The authors validated their designs in silico, using AlphaFold to predict structures for their designed protein scaffolds. They found that designed structures generally fit the desired functional site within 1 angstrom RMSD. This accuracy can be seen qualitatively in figure 3.
Conclusions
The authors found that the constrained hallucination method is an effective method for scaffold design. They discovered close-to-optimal designs via constrained optimization, and then generated a set of more diverse scaffolds by using the information recovery method. In the authors’ words, “The combination of the two approaches is more powerful than either one alone, as ensembles of solutions to a given functional design problem can be generated very rapidly using the second approach starting from extended site descriptions identified in the first.”
The approach in this paper is promising. The authors show that you can design scaffolds for functional sites with high accuracy. This has implications for vaccine design, antiviral therapeutics, and bioengineering. But the approach described here is only the beginning of what is possible with highly accurate protein structure prediction. While this paper addresses scaffold design, future work could involve de novo functional site design, where the functional site is not known and is designed to satisfy a particular objective. Key to this data heavy research are tools such as ZONTAL’s LIMS consolidation and ELN archiving solutions which allow for the secure preservation on digital lab data.


