 >
> New Work from the Creators of AlphaFold Pushes the Frontiers of Density Functional Theory

Recent advances in computational chemistry have modeled non-covalent chemical interactions like protein folding with increasing accuracy [1]. But for chemical reactions that involve bond-breaking and bond-forming, modeling is still inaccurate and computationally expensive. For chemical bonds, density-functional theory (DFT) is a field of study seeking to find a “functional” that accurately maps the electron density of an atomistic system to its energy.
In this blog, we explore a mathematically rigorous foundation that proves the existence of an exact functional, and for decades, researchers have handcrafted functionals to approximate the true functional. If we can find a better functional, we can accurately model chemical reactions at a subatomic scale, increasing our understanding of chemical interactions and opening new avenues for chemical synthesis.
DeepMind recently published a paper that introduces their own machine learning functional, which they call DM21 [1]. Their functional achieved state-of-the-art performance on several different benchmarking tasks. It was designed to satisfy some known constraints of quantum mechanics. Crucially, it allows future researchers to incorporate new constraints, allowing the model to increase in accuracy as scientific knowledge advances. Electrons have fixed properties of charge and spin (“up” or “down”). A system can’t have an overall fractional charge or fractional spin, but a region of a system can. Although many constraints such as these are known, it is difficult to manually design a functional around them. This problem is well suited for deep learning, where the constraints can be expressed as data and a functional can be trained to reproduce the energies of a system while obeying the constraints. DeepMind designed their functional with these constraints in their mind and attribute much of their success to ensuring that the constraints are satisfied.
DeepMind’s functional is shown below in Fig 1. Feature vectors of the atomistic system are calculated at certain points and are fed into a simple neural network. The outputs of the neural network at the different points are added together to get a most difficult contribution to the energy. The rest of the energy can then be calculated using mathematically exact methods to get the total energy. The algorithm was trained on a dataset of highly accurate electron densities and energies. After training, the functional can be used to predict the energy of molecules. DeepMind has made the code for their functional available for free [2].
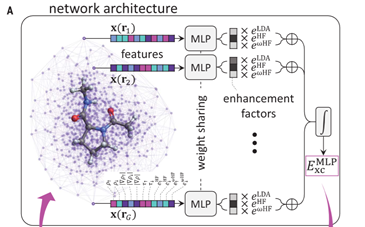
Fig 1: Deep Mind’s Functional
I expect this functional to have a significant impact in the field of computational chemistry. The combination of state-of-the-art performance and easy usability make this functional an excellent tool for computational chemistry. Highly accurate energies are important for molecular dynamics simulations, allowing more accurate investigation of chemical reactions. This network will also likely inspire further investigation of machine learning functionals and approaches to solving the DFT problem. It’s a big step toward accurate computational simulations.
Sources:
1. Highly accurate protein structure prediction with AlphaFold, John Jumper et. al, https://www.nature.com/articles/s41586-021-03819-2
2. Pushing the Frontiers of Density Functionals by Solving the Fractional Electron Problem, James Kirkpatrick, Brendan McMorrow, David H. P. Turban, Alexander L. Gaunt, James S. Spencer, Alexander G. D. G. Matthews, Annette Obika, Louis Thiry, Meire Fortunato, David Pfau, Lara Román Castellanos, Stig Petersen, Alexander W. R. Nelson, Pushmeet Kohli, Paula Mori-Sánchez, Demis Hassabis, Aron J. Cohen, Science, DOI: https://doi.org/10.1126/science.abj6511
3. Deepmind-Research GitHub page, https://github.com/deepmind/deepmind-research/tree/master/density_functional_approximation_dm21


