 >
> DiffDock – A Diffusion Model for Molecular Docking

Molecular docking is a critical task in drug design, as it involves predicting the binding structure of a small molecule ligand to a protein. Traditional methods for molecular docking rely on search-based algorithms and scoring functions to estimate the correctness of a proposed structure. However, these methods can be slow and inaccurate, especially for high-throughput workflows. In recent years, deep learning methods have been developed to tackle the problem of molecular docking by treating it as a regression problem. These methods have significantly decreased runtime compared to traditional methods, but have yet to offer substantial improvements in accuracy.
In a new research paper, a team of scientists at the Massachusetts Institute of Technology has proposed a new approach to molecular docking that frames the problem as a generative modeling problem. This means that given a ligand and target protein structure, the model learns a distribution over ligand poses. The input to the model is the 3D structure of the protein and the 3D structure of the ligand.
Figure 1: The DiffDock prediction process.
The team’s approach, called DiffDock, is a diffusion generative model (DGM) over the space of ligand poses for molecular docking. A DGM is a probabilistic model that defines a diffusion process over the degrees of freedom involved in docking, such as the position of the ligand relative to the protein, its orientation in the pocket, and the torsion angles describing its conformation. This process iteratively transforms an uninformed, noisy prior distribution over ligand poses into the learned model distribution.
One key challenge in molecular docking is the avoidance of steric clashes, which refer to the overlap of atoms between the protein and ligand that would prevent the two from binding. Traditional methods for docking often rely on heuristics to avoid steric clashes, but these can be slow and inaccurate.
DiffDock addresses this issue by defining a diffusion process over the degrees of freedom involved in docking, including the position, orientation, and torsion angles of the ligand. This allows DiffDock to sample poses that avoid steric clashes, as the progressive refinement of random poses via updates to their translations, rotations, and torsions effectively guides the ligand away from regions of the binding pocket that would result in clashes. As a result, DiffDock is able to generate high-quality ligand poses that are unlikely to be rejected due to steric clashes.
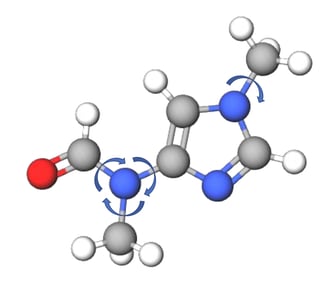
Figure 2: Torsional degrees of freedom for an example molecule.
In experiments, DiffDock significantly outperformed the previous state-of-the-art methods for molecular docking in terms of accuracy. In particular, DiffDock achieved a top-1 success rate of 38%, compared to 23% for traditional docking methods and 20% for deep learning methods. Additionally, DiffDock has fast inference times and provides confidence estimates with high selective accuracy.
In conclusion, the researchers have proposed a new approach to molecular docking that treats the problem as a generative modeling problem. This approach, called DiffDock, significantly outperforms previous methods in terms of accuracy and has fast inference times, bringing computational docking within practical reach for virtual screening in drug discovery.


